
Our Research
Overview
Proteins are the primary effectors of cells. They are highly organized in a variety of assemblies, forming the basis of well-regulated pathways and networks to precisely execute a plethora of cellular processes. Alternations of proteins expression, and their interaction networks are linked to many physiological and pathological conditions. Our main interest is to develop and apply mass spectrometry-based approaches to understand the spatial organization and dynamic regulation of protein complexes in space and time during cellular processes such as signaling and differentiation.
Developing high-throughput proteome-wide XL-MS strategies
To comprehensively understand the protein interactome, our group is developing XL-MS methods to characterize the structures and interactions of proteins within their native environments in a high-throughput manner.
Our proteome-wide XL-MS approaches allows us to characterize highly complex samples and simultaneously investigate stable and dynamic protein assemblies by capturing their residue-residue connectivities in vivo. We aim to enhance the power and scope of this technique by designing novel cross-linkers, implementing creative approaches for cross-link enrichment, developing a cutting-edge data analysis tools, and applying state-of-the-art MS technology. We integrate expertise from synthetic chemistry, analytical chemistry, mass spectrometry and informatics to advance the analytical depth, information content and precision of interactome profiling.

XL-MS workflow and its applications (image adapted from Ruwolt et al., Curr Opin Struct Biol, 2023, 10.1016/j.sbi.2023.102648).
Defining the structural interactomes of organelles, synapses, cells, and viruses
Proteins in all biological systems are highly organized in three-dimensional space, forming membrane-enclosed or membraneless compartments, signaling pathways, dynamic assemblies, and stable complexes. Many of these structures are only viable in their native environment, making them recalcitrant to traditional biochemical characterization.
Proteome-wide XL-MS offers the opportunity to capture the interactions and spatial arrangement of proteins without having to extract them from their complex biological system. Our group utilizes this technology to study the structural protein interactome of mitochondria, whole cells, and virus particles. In addition, we are interested in exploring synaptic protein interaction networks with subcompartmental specificity, defining interactomes of the presynaptic active zone, synaptic vesicles, endosomes and the postsynaptic density. These studies are instrumental for understanding synaptic plasticity and function, its adaptation to different types of stimuli and, ultimately, the molecular basis of memory and learning.
We generate comprehensive maps of the protein localizations and structural interactomes within these biological structures. These findings inform functional follow-up studies to further characterize the newly discovered protein structures, interactions, and spatially resolved networks, using approaches from structural biology, molecular biology, cell biology, neuroscience, and informatics.
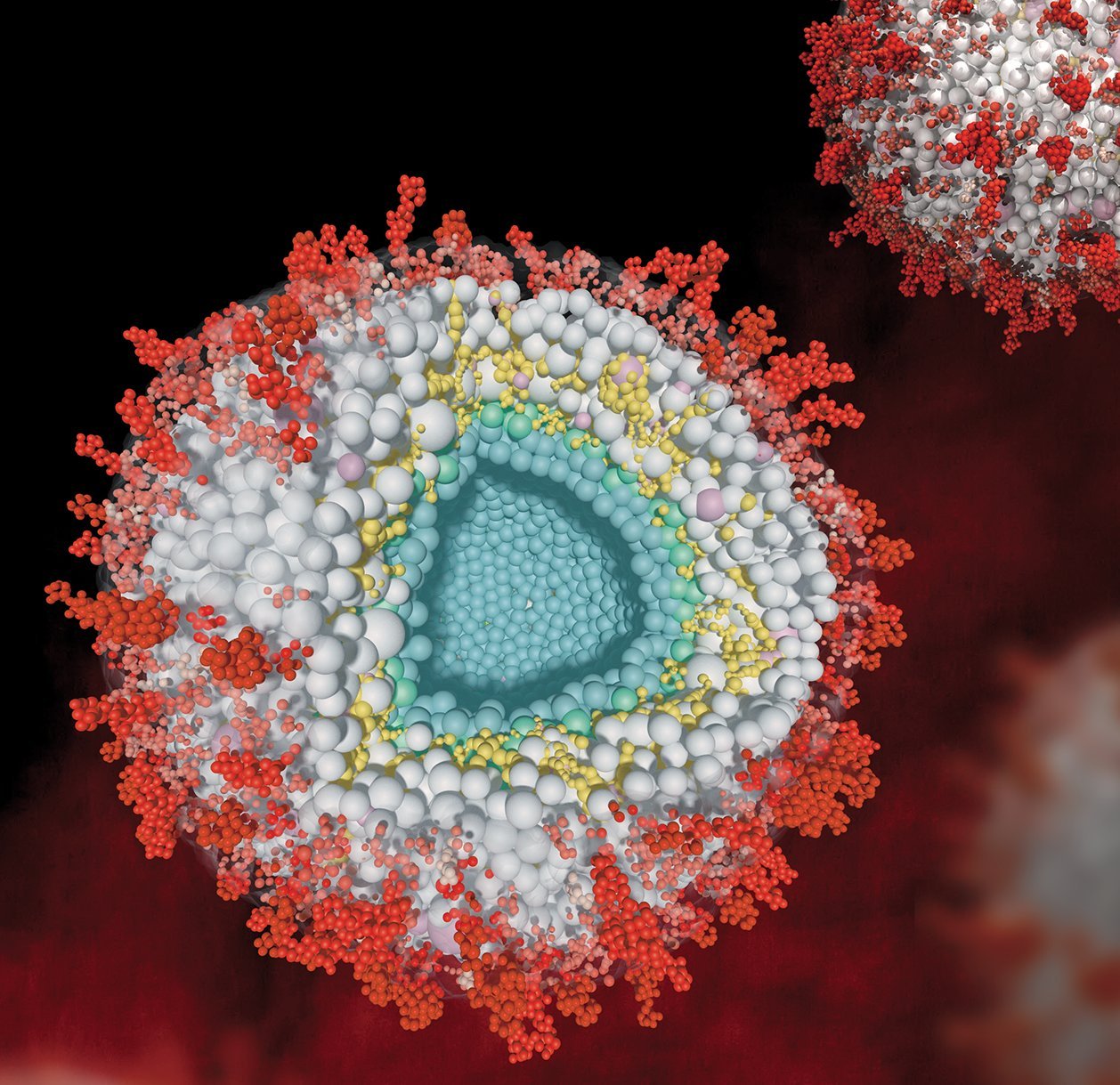
Coarse-grained model of the human cytomegalovirus particle based on XL-MS and proteomics data. Proteins are shown as beads. Their numbers are inferred from proteomics-based copy number estimates and their spatial arrangement is guided by interaction constraints from XL-MS. Pink beads represent human host proteins that are incorporated into the virion (see also Bogdanow et al., Nat Microbiol, 2023, 10.1038/s41564-023-01433-8).